R语言ComplexHeatmap复杂热图
西游东行 人气:0ComplexHeatmap|绘制单个热图介绍了单个热图绘制的内容
一 载入R包 数据
1.1 载入ComplexHeatmap包,数据
为更贴近生信使用场景,直接使用内置的基因表达数据
library(ComplexHeatmap) expr = readRDS(paste0(system.file(package = "ComplexHeatmap"), "/extdata/gene_expression.rds")) #查看数据 str(expr) expr[1:4,c(1:4,25:27)]

拿到一个新数据后,除了检查[1:4,1:4]外,也许还需要看看最后几列,另外还需要观察列名称的规律。
去除最后几列,或者只选取列名字包含cell的(TCGA数据处理中也会经常遇到)
mat = as.matrix(expr[, grep("cell", colnames(expr))])1.2 绘制最简单的热图
Heatmap(mat)
二 常见“表型”注释
文献中经常见到的就是在热图的top 或者 bottom位置添加样本的变异信息,临床信息等的注释,本节介绍如何实现以及常见的设置。
2.1读入注释文件
anno <- read.csv("anno.csv",header = T) #非真实数据,随便设置
head(anno) sample stage age#1 s1_cell01 1 56#2 s2_cell02 2 43#3 s3_cell03 2 63#4 s4_cell01 3 23#5 s5_cell02 1 8#6 s6_cell03 3 32.2添加注释,且设置颜色
2.2.1 颜色设置
1) 连续变量:指定色系,根据变量范围设置颜色范围
col_fun2 <- colorRamp2(
c(0, 50, 100), #根据值的范围设置
c("#ff7f00", "white", "#1f78b4")
)2)分类变量:直接指定颜色编码
#stage = c("1" = "red", "2" = "green", "3" = "blue" , "4" = "orange") #分类2.2.2 添加注释
使用HeatmapAnnotation函数进行注释,添加待注释的内容
ha <- HeatmapAnnotation(
age = anno$age,
stage = anno$stage,
col = list(
age = col_fun2 , #连续
stage = c("1" = "red", "2" = "green", "3" = "blue" , "4" = "orange") #分类
)
)1)注释位置
#指定注释位置 ,示例为top_annotation,此外可选 bottom_annotation ,right_annotation ,left_annotation
Heatmap( mat, top_annotation = ha )

热图上面注释样本的临床等信息,实现!
2) 指定多个注释位置
当需要注释的内容较多时候,可以选择在不同的位置。需要预先根据待注释的位置进行指定
column_ha <- HeatmapAnnotation( bar1 = anno_barplot(runif(24)) ) row_ha <- rowAnnotation( bar2 = expr$chr ) Heatmap( mat, show_row_names = F , #cluster_rows = F , top_annotation = ha , bottom_annotation = column_ha, #对应的注释 right_annotation = row_ha )

其他常用调整的函数
#cluster_rows/columns :是否进行聚类
#show_column/row_dend :是否显示聚类树
#column/row_dend_side :聚类图绘制的位置
#column_dend_height/row_dend_widht :聚类树的高度 和 宽度
三 添加“块”注释
常见的是根据聚类(kmeans等)或者 先验知识 分为几个簇,然后对簇进行注释。
3.1 k-means指定K个数
1)样本设置分为4组,基因分为3组,同时设置每个“簇”的颜色和标签
set.seed(1234)
Heatmap(mat,
top_annotation = HeatmapAnnotation(foo = anno_block(gp = gpar(fill = 1:4),
labels = c("group1", "group2", "group3", "group4"),
labels_gp = gpar(col = "white", fontsize = 10))),
column_km = 4, # 列分为4个k
left_annotation = rowAnnotation(foo = anno_block(gp = gpar(fill = 2:4),
labels = c("group1", "group2", "group3"),
labels_gp = gpar(col = "white", fontsize = 10))),
row_km = 3, #
show_row_names = F
)
2)设置 text的颜色
Heatmap(mat,
top_annotation = HeatmapAnnotation(foo = anno_block(gp = gpar(fill = 1:4),
labels = c("group1", "group2", "group3", "group4"),
labels_gp = gpar(col = "white", fontsize = 10))),
column_km = 4,
left_annotation = rowAnnotation(foo = anno_block(gp = gpar(fill = 2:4),
labels = c("group1", "group2", "group3"),
labels_gp = gpar(col = "white", fontsize = 10))),
row_km = 3,
show_row_names = F ,
row_title_gp = gpar(
col = rainbow(5)[2:4],
font = 1:3
),
row_names_gp = gpar(
col = rainbow(5)[2:4],
fontsize = 10:12
),
column_title_gp = gpar(
fill = rainbow(5)[1:4],
alpha = 0.5
),
column_names_gp = gpar(
col = rainbow(5)[1:4]
)
)
关于颜色可选#rainbow,heat.colors,terrain.colors,topo.colors,cm.colors
3.2 先验知识知道样本分为几个簇
指定样本添加列注释,假设mat中的24个样本,已知是分别为10个,10个 和4个的三组 。
实际应用中可以根据 年龄段,性别,临床分析,预后评分等指标进行的分组。
split = c( rep(c("A","B"),10) , rep("C",4) )
ha = HeatmapAnnotation(foo = anno_block(gp = gpar(fill = 2:6), labels = c("AA","BB","CC") ))
col_fun = colorRamp2(c(0, 5, 10, 20), c("white", "cornflowerblue", "yellow", "red"))使用column_split 函数即可按照指定拆分
Heatmap(mat,
name = "mat_cluster",
column_split = split,
top_annotation = ha,
cluster_rows = T,
cluster_columns = F,
#rect_gp = gpar(col="white"), #添加白色格子线
column_title = NULL)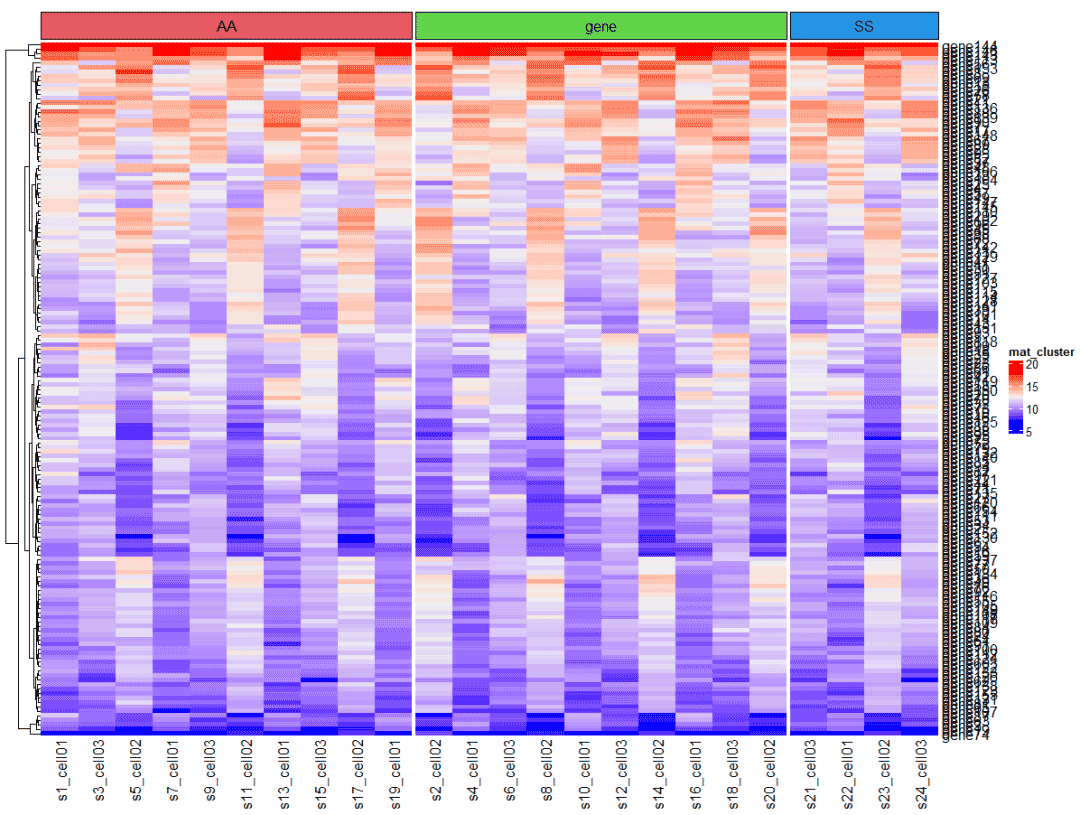
3.3 根据富集结果添加行注释
文献中经常见到 一些基因富集的通路作为 行注释的图,怎么实现呢?
1)自定义通路结果(也可以是其他想展示的内容)
group <- list( A = "Cell cycle", B = "Mismatch repair", C = "DNA replication" )
2)添加空白注释
ha = rowAnnotation(
foo = anno_empty(
border = FALSE,
# 计算空白注释的宽度
width = max_text_width(unlist(group)) + unit(4, "mm"))
)3)通过向量拆分对应的行和列
Heatmap(mat, name = "mat",
#cluster_rows = T,
show_row_names = F,
right_annotation = ha,
row_split = c( rep(c("A","B"),30) , rep("C",95) ) ,
column_split = rep(c("C", "D"), 12))4)添加注释块 以及 注释文本
for(i in 1:3) {
decorate_annotation(
"foo",
# 选择热图块
slice = i, {
# 添加颜色框
grid.rect(
x = 0,
width = unit(2, "mm"),
gp = gpar(
fill = rainbow(3)[i],
col = NA
),
just = "left"
)
# 绘制文本
grid.text(
group[[i]],
x = unit(4, "mm"),
gp = gpar(
col = rainbow(3)[i]
),
just = "left")
})
}
需要注意的是 这里需要对应好,各位有更好的方法希望不吝告知。
四 目标基因分析
4.1 标签展示目标基因
使用anno_mark() 函数展示目标基因,至少需要两个参数,通过at 提供原始数据矩阵的索引,labels 为相应的文本标记。
1)读取待展示的基因名称,也可以是geneList的向量
name <- read.table('name.txt', header = T, check.names = FALSE)
head(name)
# gene#1 gene3#2 gene53#3 gene6#4 gene78#5 gene7#6 gene92)获取目标基因对应的矩阵位置;
genelist <- name$gene index <- which(rownames(mat) %in% genelist) #得到对应的文本标签; labs <- rownames(mat)[index]
3)使用labels_gp调整字体大小;
lab2 = rowAnnotation(foo = anno_mark(at = index,
labels = labs,
labels_gp = gpar(fontsize = 8),
lines_gp = gpar()))标签展示目标基因
Heatmap(mat, name = "mat",
cluster_rows = T,
right_annotation = lab2,
row_names_side = "right",
show_row_names = F,
row_names_gp = gpar(fontsize = 4))
4.2 绘制目标基因热图
大部分热图存在基因太多的情况,重点展示目的基因 。
heatmap4 <- Heatmap( mat, name = "expression" ) heatmap
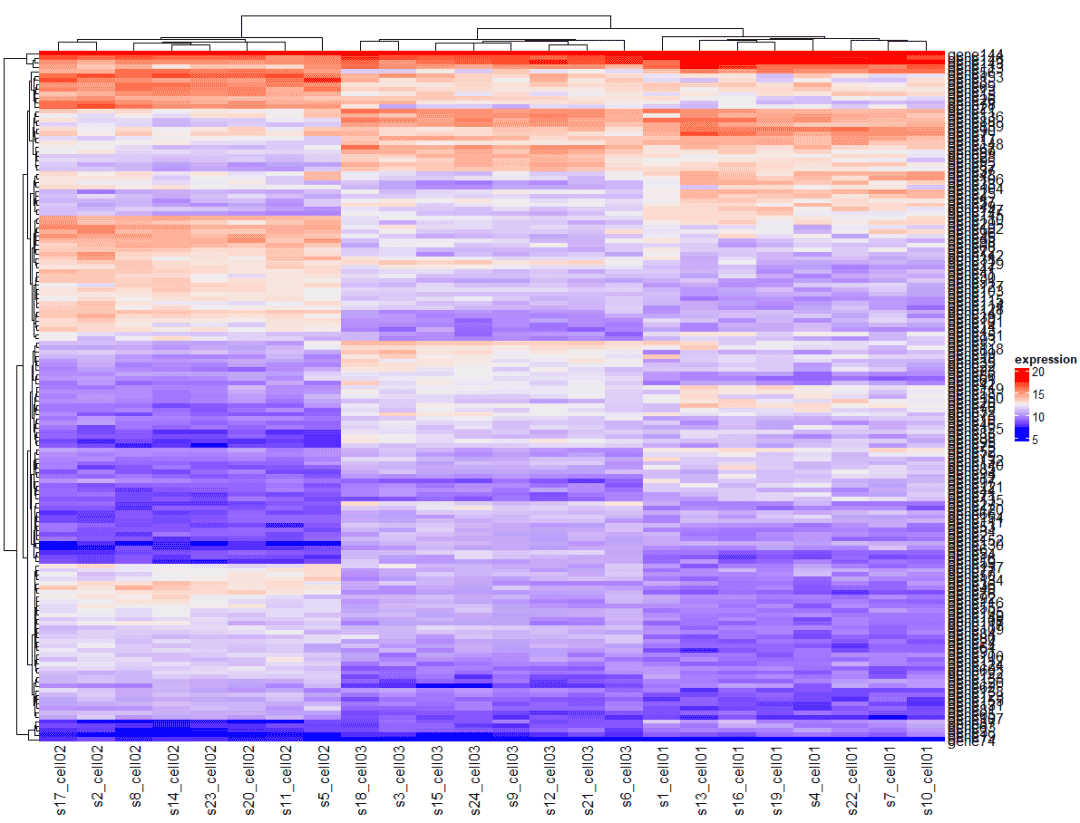
4.2.1 在总图中提取出来目标基因的热图,颜色与大图一致
提取目的基因所在的位置进行绘制
heatmaph4[c(1,5,6,8,9,80,144,74),]

这种方式是在总的热图中直接提取目的基因的部分,热图的颜色与总的热图一致。
4.2.2 提取基因数据重新绘制热图
labs2 <- c("gene1", "gene5", "gene6", "gene8", "gene9", "gene80" ,"gene144", "gene74")
mat2 <- as.data.frame(mat) %>%
rownames_to_column("gene") %>%
filter( gene %in% labs2 ) %>%
column_to_rownames("gene")
Heatmap(mat2)
注意区别
参考资料:
https://jokergoo.github.io/ComplexHeatmap-reference/book/a-single-heatmap.html
注释及基因文件均为随便设置的,可根据数据情况自行更改
加载全部内容