python PyVCF处理VCF格式
陈光辉_花生所 人气:0引言
vcf文件的全称是variant call file,即突变识别文件,它是基因组工作流程中产生的一种文件,保存的是基因组上的突变信息。通过对vcf文件进行分析,可以得到个体的变异信息。嗯,总之,这是很重要的文件,所以怎么处理它也显得十分重要。它的文件信息如下:
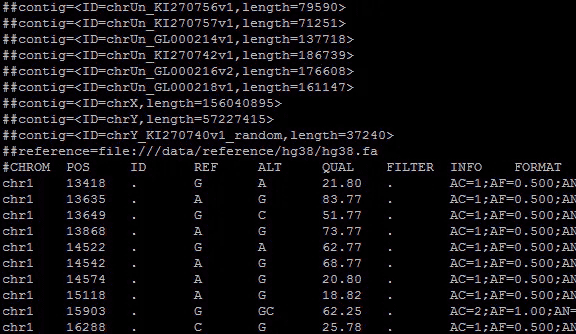
文件的开头是一堆以“##”开始的注释行,包含了文件的基本信息。然后是以“#”开头的一行,共9+n个部分,前九部分标注的是后面行每部分代表的信息,相当于表头。后面部分是样本名称,可以有多个。注释行结束后是具体的突变信息,每一行分为9+n个部分,每部分之间用制表符(‘\t’)分隔。
通常处理vcf文件时,在读取,处理阶段总是会写很多重复代码,核心的任务代码很少。当然,如果仅仅是找位点的CHROM,POS,ID,REF,ALT,QUAL这几个参数时,这样做也可以。因为vcf格式规范,这几个参数的结构相对简单。但是如果处理头文件信息,或者处理INFO,FORMAT参数时,要写比较复杂的正则表达式,这样做不仅繁琐,而且容易出错。
Python的PyVCF库解决了这个问题,它通过正则表达式把vcf文件信息转换成结构化的信息,简化了vcf文件的处理过程,方便后续提取相关参数及处理。
PyVCF库的安装
cmd界面
pip install PyVCF
或者从https://github.com/jamescasbon/PyVCF网站上下载安装包,自行安装。
PyVCF库的导入
import vcf
PyVCF库的名字为vcf,导入之后可以使用其方法对vcf文件做处理。
PyVCF库详细介绍
使用实例:
>>> import vcf >>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz') >>> for record in vcf_reader: print recordRecord(CHROM=chr1, POS=10146, REF=AC, ALT=[A])Record(CHROM=chr1, POS=10347, REF=AACCCT, ALT=[A])Record(CHROM=chr1, POS=10439, REF=AC, ALT=[A])Record(CHROM=chr1, POS=10492, REF=C, ALT=[T])Record(CHROM=chr1, POS=10583, REF=G, ALT=[A])
调用vcf.Reader类处理vcf文件,vcf文件信息就被保存到vcf_reader中了。它是一个可迭代对象,它的迭代元素都是一个_Record对象的实例,保存着非注释行的一行信息,即变异位点的具体信息。通过它,我们可以很轻易地得到位点的详细信息。
_Record对象------位点信息的储存形式
class vcf.model._Record(CHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO, FORMAT, sample_indexes, samples=None)
_Record是vcf.model中的一个对象,除了它还有_Call,_AltRecord等对象。它的基本属性为CHROM,POS,ID,REF,ALT,QUAL,FILTER,INFO,FORMAT,也就是vcf中的一行位点信息。接下来对这些属性一一说明:
CHROM:染色体名称,类型为str。
POS:位点在染色体上的位置,类型为int。
ID:一般是突变的rs号,类型为str。如果是‘.’,则为None。
REF:参考基因组在该位点上的碱基,类型为str。
ALT:在该位点的测序结果。是_AltRecord类的子类实例的列表。类型为list。_AltRecord类有4个子类,代表了突变的几种类型:如snp,indel,structual variants等。所有的实例都可以进行比较(仅限于相等的比较,没有大于小于之说),部分子类没有实现str方法,也就是说不能转成字符串。
QUAL:该位点的测序质量,类型为int或float。
FILTER:过滤信息。将FILTER列按分号分隔形成的字符串列表,类型为list。如果未给出参数则为None。
INFO:该位点的一些测试指标。将‘=’前的参数作为键,后面的参数作为值,构建成的字典。类型为dict。
FORMAT:基因型信息。保存vcf的FORMAT列的原始形式,类型为str。
>>> for record in vcf_reader:
print type(record.CHROM), record.CHROM
print type(record.POS), record.POS
print type(record.ID), record.ID
print type(record.REF), record.REF
print type(record.ALT), record.ALT
print type(record.QUAL), record.QUAL
print type(record.FILTER), record.FILTER
print type(record.INFO), record.INFO
print type(record.INFO['BaseQRankSum']), record.INFO['BaseQRankSum']
print type(record.FORMAT), record.FORMAT
<type 'str'> chr1<type 'int'>
234481<type 'NoneType'> None<type 'str'>
T<type 'list'> [A]<type 'float'> 2025.77<type 'NoneType'>
None<type 'dict'> {'ExcessHet': 3.0103, 'AC': [1],
'BaseQRankSum': -2.743, 'MLEAF': [0.5], 'AF': [0.5],
'MLEAC': [1], 'AN': 2, 'FS': 2.371, 'MQ': 42.83,
'ClippingRankSum': 0.0, 'SOR': 0.972, 'MQRankSum': -2.408,
'ReadPosRankSum': 1.39, 'DP': 156, 'QD': 13.07}<type 'float'>
-2.743<type 'str'>GT:AD:DP:GQ:PL
除了这些基本属性之外,_Record对象还有一些其他属性:
samples:把FORMAT信息作为键,后面对应的信息做为值,构建成的字典(CallData对象),以及sample名称,这两个值组成一个Call对象,共同构成samples的一个元素。这样就把sample和基因型信息给关联起来,按下标访问每一个Call对象。samples类型为list。
start:突变开始的位置
end:突变结束的位置
alleles:该位点所有的可能情况,由REF和ALT参数组成的列表(REF类型是str,ALT参数是_AltRecord对象的子类实例),类型是list。
>>> for record in vcf_reader: print record.samples, '\n', record.samples[0].sample, '\n', record.samples[0]['GT'] #按下标访问Call,按.sample访问sample,按键访问FORMAT对应信息 print record.start, record.POS, record.end print record.REF, record.ALT, record.alleles #注意G没有引号,它是_AltRecord对象 [Call(sample=192.168.1.1, CallData(GT=0/1, AD=[39, 14], DP=53, GQ=99, PGT=0|1, PID=13116_T_G, PL=[449, 0, 2224]))] 192.168.1.10/113115 13116 13116T [G] ['T', G]
_Record对象方法:
- 对象之间比较大小方法:根据染色体名称和位置信息比较。Python3只实现了‘=’和‘<’的比较。
- 迭代方法:对samples里的元素进行迭代。
- 字符串方法:只返回CHROM,POS,REF,ALT四列信息。
- genotype(name)方法,和samples按下标访问不同,这个方法提供按sample名称进行访问的功能。
- add_format(fmt), add_filter(flt), add_info(info, value=True):给相应的属性添加元素。
- get_hom_refs():拿到samples中该位点未突变的所有sample,返回列表。
- get_hom_alts():拿到samples中该位点100%突变的所有sample,返回列表。
- get_hets():拿到samples中该位点基因型为杂合的所有sample,返回列表。
- get_unknown():拿到samples中该位点基因型未知的所有sample,返回列表。
>>> record = next(vcf_reader)
>>> record2 = next(vcf_reader)
>>> print record > record2
#按染色体名称和位置进行比较False
>>> for i in record:
#按samples列表进行迭代
print i
Call(sample=192.168.1.1,
CallData(GT=0/1, AD=[18, 11],
DP=29, GQ=99, PL=[280, 0, 528]))
>>> print str(record)
#字符串方法Record(CHROM=chr1, POS=10492, REF=C, ALT=[T])
>>> print record.genotype('192.168.1.1')
#按sample名字进行访问
Call(sample=192.168.1.1,
CallData(GT=0/1, AD=[39, 14], DP=53, GQ=99, PGT=0|1, PID=13116_T_G, PL=[449, 0, 2224]))
_Record对象还有很多有用的方法属性:
num_called:该位点已识别的sample数目。
call_rate:已识别的sample数目占sample总数的比例。
num_hom_ref,num_hom_alt,num_het,num_unknown:四种基因型的数量
aaf:所有sample等位基因的频率(即除开REF),返回列表。
heterozygosity:该位点的杂合度,0.5为杂合突变,0为纯合突变。
var_type:突变类型,包括‘snp’,‘indel’,‘sv’(structural variant),‘unknown’。
var_subtype:更加细化的突变类型,如‘indel’包括‘del’,‘ins’,‘unknown’。
is_snp,is_indel,is_sv,is_transition,is_deletion:判断突变是不是snp,indel,sv,transition,deletion等等。
>>> record = next(vcf_reader) >>> print recordRecord(CHROM=chr1, POS=13118, REF=A, ALT=[G]) >>> print record.samples #只有一个sample [Call(sample=192.168.1.1, CallData(GT=0/1, AD=[41, 13], DP=54, GQ=99, PGT=0|1, PID=13116_T_G, PL=[449, 0, 2224]))] >>> record.num_called1 >>> record.call_rate1.0 >>> record.num_hom_ref0 >>> record.aaf[0.5] >>> record.num_het1 >>> record.heterozygosity0.5 >>> record.var_type'snp' >>> record.var_subtype'ts' >>> record.is_snpTrue >>> record.is_indelFalse
Reader对象------处理vcf文件,构建结构化信息
class Reader(fsock=None, filename=None, compressed=None, prepend_chr=False, strict_whitespace=False, encoding='ascii')
在读vcf文件时,总共有六个参数可供选择,如上图所示。
fsock:目标文件的文件对象,可以用open(文件名)得到这个文件对象。
filename:文件名,当fsock和filename同时存在时,优先考虑fsock。
compressed:是否要解压,不提供参数时由程序自行判断(以文件名是否以.gz结尾判断是否需要解压)。
prepend_chr:在保存染色体名称时,是否加前缀‘chr’,默认不加,如果vcf文件的染色体名称本来没有前缀‘chr’,可设置为True,自动加上。
strict_whitespace:是否严格以制表符‘\t’分隔数据。True则表示严格按制表符分,False表示可以夹杂空格。
encoding:文件编码。
>>> vcf_reader = vcf.Reader(open('vcf/test/example-4.0.vcf', 'r'))
#fsock
>>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz')
#filename
头文件信息主要保存在Reader对象的属性中,包括alts,contigs,filters,formats,infos,metadata。
alts使用实例:
>>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz')
>>> vcf_reader.altsOrderedDict([('NON_REF', Alt(id='NON_REF', desc='Represents any possible alternative allele at this location'))])
#字典类型
>>> vcf_reader.alts['NON_REF'].id'NON_REF'
>>> vcf_reader.alts['NON_REF'].desc'Represents any possible alternative allele at this location'
其他的属性用法类似。
Reader对象实现了两个方法:
next():获得下一行的数据,也就是返回下一个_Record对象。可以显式调用next()得到下一行数据,也可以直接迭代Reader对象,它会自动调用next()函数以获得下一行数据。
fetch(chrom,start=None,end=None):返回chrom染色体从start+1到end坐标的所有突变位点。不给end,就返回chrom染色体从start+1到末尾的所有突变位点;
start和end都不给,就返回chrom染色体所有的突变位点。这个方法需要用另一个第三方Python模块pysam来建立文件索引,如果没有安装这个模块,会导致错误。
另外,使用这个方法之后,它会将对象的可迭代范围改成fetch()得到的突变位点,所以用这个方法,原来的迭代进度就失效了。
>>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz') >>> vcf_reader.next()<vcf.model._Record object at 0x0000000003ED8780 >>>> record = vcf_reader.next() >>> print recordRecord(CHROM=chr1, POS=10347, REF=AACCCT, ALT=[A]) >>> for record in vcf_reader: print recordRecord(CHROM=chr1, POS=10439, REF=AC, ALT=[A])Record(CHROM=chr1, POS=10492, REF=C, ALT=[T])
这个库还有一个Writer对象,在此就不详细介绍了,因为大部分对vcf文件的处理都可以用上面两个对象的知识搞定。
综合使用:
#!/usr/bin/env python
# -*- coding: utf-8 -*- import vcf
# 导入PyVCF库
filename = r'D:\test\example.hc.vcf.gz'
vcf_reader = vcf.Reader(filename=filename)
# 调用Reader对象处理vcf文件
for record in vcf_reader:
# 迭代Reader对象,返回的是_Record对象
# record是_Record对象
print record.CHROM, record.POS, record.ID, record.ALT
if record.is_snp:
# 判断是否是snp
print "I'm a snp"
elif record.var_type != 'sv':
#和 elif record.is_sv:等价
print "I'm not a sv"
if record.heterozygosity == 0.5:
# 判断是否为杂合突变
print "I'm a heterozygous mutation"
... ...
这个库实现的所有功能,都可以自己写代码实现,而且实现方法比较简单。之所以要用这个库来处理vcf文件,是因为这个库考虑的东西可能比我们自己了解的更多,其实现也可能比我们自己的代码更加完备合理。
还有,重复造车总归是不好的。
加载全部内容